
18244218588
蛋白多序列比對(Multiple Sequence Alignment, MSA)是一種將三個或更多的蛋白質序列進行排列的技術,以便zuì大程度地識別出它們之間的相似和差異。分析蛋白多序列比對結果可以揭示蛋白家族成員之間的進化關系、結構域、功能位點以及其它生物學上的重要信息。
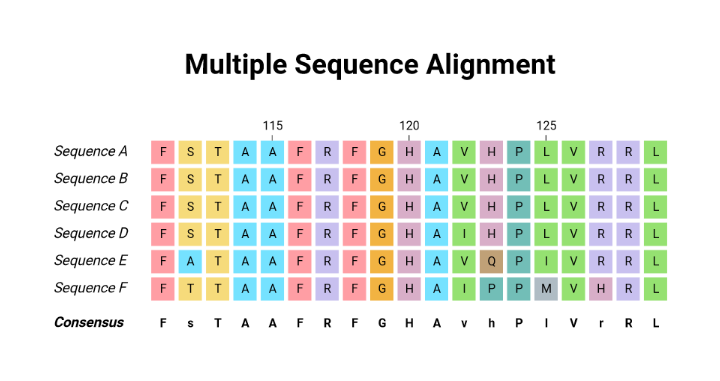
圖1. 蛋白多序列比對
分析MSA的結果涉及多個步驟,包括比對質量的評估、保守性分析、進化關系的推斷以及功能位點的預測等:
一、比對質量評估
檢查比對的一致性:觀察是否存在大量的插入或缺失區域,以及是否有蛋白質序列與其他序列顯著不同,這些都可能是比對質量不高的信號。
使用評分系統:利用如SP-score或其他比對質量評分工具,對比對的整體質量進行量化評估。
可視化工具:使用如Jalview、MAFFT的可視化界面,幫助直觀地檢查比對的準確性和一致性。
二、保守性分析:
識別保守區域:查找在多個序列中高度保守的氨基酸殘基,這些區域通常與蛋白質的功能密切相關。
計算保守性分數:使用工具如Consurf,根據氨基酸在進化過程中的變化頻率,給每個位置的保守性打分。
保守性圖譜:生成保守性圖譜,直觀展示每個位置的保守性,用于預測功能域或活性位點。

圖2. 蛋白多序列比對的序列標志表示
三、進化關系推斷:
構建系統發育樹:使用MSA結果構建系統發育樹,推斷不同蛋白質序列之間的進化關系。
分析進化分支:通過分析系統發育樹的分支結構,可以推測蛋白家族的進化歷史以及功能分化。
識別同源序列:通過比對和系統發育分析,識別出與已知功能蛋白質相似的序列,推測其潛在功能。
四、功能位點預測
關鍵位點識別:基于保守性分析,識別可能的功能位點或活性中心。
結構預測:如果可能,結合蛋白質三維結構信息,進一步驗證預測的功能位點的準確性。
文獻驗證:將分析結果與已發布的研究結果進行比較,驗證預測的功能位點或結構域的相關性。
通過上述步驟,可以從多序列比對的結果中提取有價值的生物學信息,為蛋白質的功能研究以及進化分析提供重要的線索。
請輸入賬號
請輸入密碼
請輸驗證碼
以上信息由企業自行提供,信息內容的真實性、準確性和合法性由相關企業負責,化工儀器網對此不承擔任何保證責任。
溫馨提示:為規避購買風險,建議您在購買產品前務必確認供應商資質及產品質量。



